Behind every approved drug is a decade or more of scientific rigor, clinical precision and regulatory complexity. This process, which takes a compound from the lab to pharmacy shelves, is one of the most demanding in science and industry. The path from identifying a promising molecule to delivering a commercial drug is long, costly and fraught with risk, but it is also essential to medical progress.
Sponsored Article
From Molecule to Market - Navigating the Drug Development Process

Published May 26, 2025 18:42

An Overview of the Drug Development Journey - From Discovery to Commercialization
The development of a new drug, particularly in biologics process development, is a rigorous, multi-phase process that transforms scientific innovation into a safe, effective, and marketable therapy. Each stage plays a vital role in ensuring that a drug meets regulatory expectations while addressing unmet medical needs.
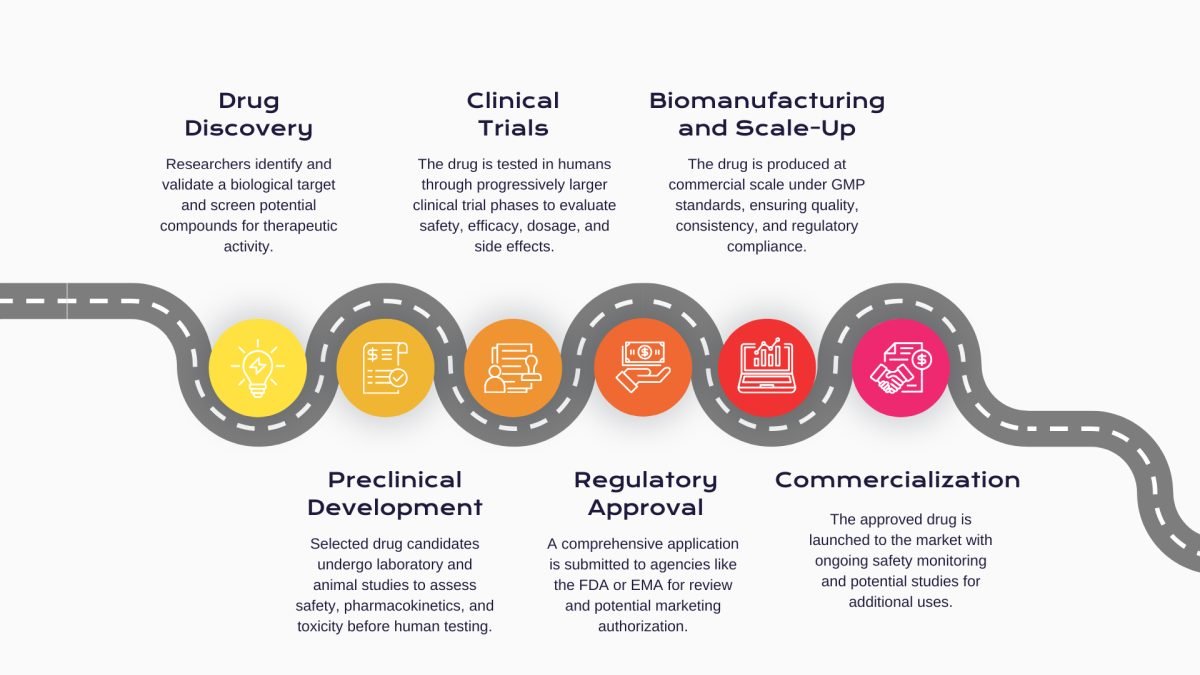
The journey begins in the laboratory, where scientists identify a biological target, usually a protein or gene that plays a key role in the disease process.High-throughput screening technologies are then used to evaluate thousands of molecular candidates for their ability to interact with the target. At this stage, emphasis is placed on understanding the mechanism of action, optimizing selectivity, and assessing early indicators of therapeutic potential. Only a fraction of compounds demonstrate the necessary attributes to advance to the next phase.
Once promising candidates are identified, they enter the preclinical phase, which involves a series of in vitro and in vivo studies designed to evaluate safety and biological behavior. Researchers assess pharmacokinetics, pharmacodynamics and conduct toxicology bioassays to identify any adverse effects. These studies are essential for establishing a safe starting dose for human trials. The data generated are compiled into an Investigational New Drug (IND) application, a regulatory requirement for initiating clinical trials.
Clinical Trials - Critical Phases in Drug Development
After submitting the IND, the sponsor must wait 30 calendar days before initiating any clinical trials. During this time, FDA has an opportunity to review the IND for safety to assure that research subjects will not be subjected to unreasonable risk.If the FDA identifies any safety concerns, it may place the study on clinical hold, preventing it from beginning until the issues are resolved. This critical safety checkpoint marks the transition from laboratory science to human research and sets the stage for a highly structured, phased clinical development process.
Phase I trials are the first step in testing a drug in humans, typically involving 20 to 100 healthy volunteers or, in the case of high-risk therapies, patients with the target disease. The primary objective is to assess the drug's safety profile, determine a safe dosage range, and gather pharmacokinetic and pharmacodynamic data. These trials are usually conducted in a controlled inpatient setting to allow close monitoring for any adverse effects. Although therapeutic benefit is not the goal at this stage, Phase I trials provide the foundational data necessary to progress further in clinical development.
Phase II serves as a critical decision point only candidates that demonstrate meaningful clinical benefit with an acceptable safety margin are advanced to the next stage. Population of that clinical trials involve several hundred patients and aim to evaluate the drug’s efficacy for a specific indication while continuing to monitor its safety. These trials are typically randomized and controlled, sometimes comparing different doses of the investigational product or comparing it to a placebo or existing therapy.
Phase III trials are pivotal studies involving large patient populations, often ranging from several hundred to several thousand participants, conducted at multiple sites across various geographies. The primary goals are to confirm efficacy, monitor for less common side effects, and collect the robust safety and effectiveness data needed for regulatory approval. These trials are often double-blinded and include active comparators to demonstrate how the new therapy performs against current standards of care. Phase III results form the core clinical component of the New Drug Application (NDA) or Marketing Authorization Application (MAA) and are essential in determining a drug's benefit-risk profile.
Throughout each phase, clinical trials are conducted under strict ethical and scientific standards. Good Clinical Practice (GCP) guidelines ensure that patient welfare is prioritized, data is collected rigorously, and trials are executed in a transparent and reproducible manner. These principles not only protect participants but also build the trust and reliability that the healthcare community and regulators require before a drug can reach the market.
Regulatory Pathways and Approval Processes in the Pharma Industry
Bringing a new drug to market involves more than scientific achievement; it requires successfully navigating the intricate regulatory landscapes that govern the approval of medicines. U.S. Food and Drug Administration (FDA) and the European Medicines Agency (EMA) set the global standards for ensuring that new therapies are safe, effective, and of high quality. While both institutions share similar goals, their approval processes follow different pathways, each with detailed and carefully structured steps.
The FDA Approval Process
In the United States, the FDA oversees the regulatory approval of new drugs through a structured sequence of pre- and post-submission activities. The process begins with pre-submission engagement, typically in the form of a Pre-NDA (New Drug Application) meeting. This allows the sponsor to discuss with FDA reviewers key elements of the upcoming application, such as clinical trial results, CMC (Chemistry, Manufacturing, and Controls) data, and labeling proposals. For therapies addressing serious or life-threatening conditions, the FDA offers expedited pathways such as Fast Track, Breakthrough Therapy Designation, Accelerated Approval, and Priority Review, all aimed at reducing development timelines.
Once pivotal Phase III data are available and the development program is complete, the sponsor compiles the NDA. This submission includes comprehensive datasets from preclinical and clinical trials, manufacturing protocols, analytical validation, proposed labeling, and a detailed risk management strategy (often in the form of a Risk Evaluation and Mitigation Strategy, or REMS). The NDA is formatted using the Common Technical Document (CTD) structure to ensure global compatibility and ease of review.
Upon receipt of the NDA, the FDA conducts a 60-day filing review to ensure the application is complete. If accepted, the FDA assigns a PDUFA (Prescription Drug User Fee Act) date, which defines the timeline for decision-making: 10 months for standard review and 6 months for priority applications. During the review phase, multidisciplinary teams assess clinical safety and efficacy, quality of manufacturing processes, and benefit-risk balance. The FDA may conduct inspections of manufacturing facilities to confirm GMP compliance, review batch records, and assess quality systems.
If the evaluation concludes positively, the FDA issues an Approval Letter, which may include post-marketing requirements. If deficiencies are identified, the agency sends a Complete Response Letter outlining the issues that must be resolved before approval can be granted. After approval, sponsors must maintain regulatory obligations through pharmacovigilance, post-marketing studies, and supplemental filings for any changes to the product or its usage.
The EMA Approval Process
In Europe, the regulatory pathway for centralized drug approval is managed by the EMA, whose procedures ultimately result in marketing authorization across all EU member states. As with the FDA, the EMA encourages early dialogue between sponsors and regulators. This occurs through Scientific Advice meetings, where development plans, clinical protocols, and CMC strategies can be discussed and refined in alignment with regulatory expectations. For medicines that address unmet medical needs or offer major therapeutic advancements, sponsors may apply for the EMA’s PRIME (PRIorityMEdicines) designation, which facilitates closer guidance and accelerated assessment.
The formal process begins with the submission of a Marketing Authorization Application (MAA), structured according to the CTD format and including data on nonclinical studies, clinical trials, manufacturing, labeling, and a Risk Management Plan (RMP). Once the MAA is received, the EMA initiates a validation period to confirm administrative completeness. Upon validation, the review process begins with the Committee for Medicinal Products for Human Use (CHMP), which leads the scientific evaluation.
The standard EMA review timeline is 210 days, interrupted once for a "clock stop" period that allows the sponsor to address any questions raised by the CHMP. The review evaluates the same three core pillars as the FDA: product efficacy, safety, and quality. If necessary, the EMA may conduct GMP inspections at manufacturing sites within or outside of Europe. Once the scientific review is complete, the CHMP issues a recommendation: positive or negative. A positive opinion is forwarded to the European Commission, which grants final, legally binding approval valid throughout the EU.
Following authorization, EMA oversight continues. Companies are required tomaintain robust pharmacovigilance systems, submit Periodic Safety Update Reports (PSURs), and conduct additional studies as agreed during the review. Changes to the approved product, such as new indications, label updates, or manufacturing changes, require formal variation applications.
The Role of Strategic Partnerships and CDMO Companies in Drug Development
In the high-stakes landscape of biopharmaceutical innovation, no company succeeds in isolation. Strategic partnerships with CDMOs play an increasingly central role in the drug development process. These collaborations enable sponsors to leverage specialized capabilities, including biologics process development, expand development bandwidth, and accelerate time-to-market, all while maintaining regulatory rigor. From early-phase development through commercial manufacturing and product launch, CDMOs act as critical enablers of biopharma success.
CDMO companies bring a unique blend of scientific expertise, technological infrastructure, and regulatory acumen that many emerging biotech firms or lean pharmaceutical sponsors may lack in-house. Whether it’s developing a robust cell line, scaling a downstream purification process, or validating analytical methods, CDMOs provide the technical depth needed to translate innovation into manufacturable products. Importantly, their familiarity with FDA and EMA expectations reduces regulatory risk and facilitates smoother transitions through the development pipeline.
As the investigational drug advances toward late-stage development and regulatory submission, the value of CDMOs becomes even more pronounced. In preparation for regulatory approval, sponsors must ensure that all manufacturing processes - both for the drug substance (DS) and drug product (DP) - are validated according to Good Manufacturing Practices (GMP). CDMOs with established GMP-compliant infrastructure and global regulatory experience can rapidly implement manufacturing protocols that meet the necessary standards. Their facilities are routinely audited and often pre-inspected by regulatory agencies, expediting approval timelines and mitigating compliance issues.
Once approval is secured, the product enters the manufacturing and scale-up phase. This is a critical inflection point where speed, reliability, and consistency become paramount. CDMOs manage the transition from clinical-scale to commercial-scale manufacturing, ensuring that technology transfer is seamless and that processes are scalable and reproducible. Their teams validate batch consistency, conduct stability testing, and implement real-time release strategies to meet both supply chain demand and global regulatory obligations. This is especially important for biologics, where process variability can significantly impact product quality.
In addition to manufacturing, CDMOs are key players in commercialization. They support product launch by ensuring sufficient inventory levels, packaging in compliance with regional requirements, and maintaining the cold chain for temperature-sensitive biologics. Their logistical expertise and supply chain networks ensure timely global distribution.
Post-approval, the sponsor’s responsibilities extend far beyond production. Pharmacovigilance, post-marketing studies, and regulatory maintenance require ongoing resources. Here again, strategic CDMO partnerships offer long-term value. Many CDMOs provide lifecycle management services such as reformulation, new delivery systems, or manufacturing support for expanded indications. They also assist with supplemental regulatory submissions, documentation updates, and risk mitigation strategies based on real-world data, all of which contribute to sustained product success.
Conclusion
Drug development is far more than a sequence of technical steps.Each phase (scientific, clinical, regulatory, and commercial) must be executed with scientific integrity and operational excellence.Through a combination of rigorous science, comprehensive regulatory engagement, and the operational power of CDMO partnerships, companies can transform groundbreaking discoveries into globally available treatments.
Topics
Sponsored Article